Institutes & Centers
Institute for Biomedicine
Institute for Biomedicine
- Deutsch
- English
- Italiano
Our starting point is always the individual and their unique characteristics, then comes the laboratory and finally we go back to people: this is how our research work is undertaken. We study how genetic heritage, the environments in which we live and our lifestyles affect the appearance and development of diseases as well as how these factors influence our health.
1 - 7






Facts
& Figures
77
Staff members
39 with a PhD degree
46
Ongoing projects
28 of them with international partners
56
Journal publications
8
Contributions in conference proceedings
3
Laboratories and facilities
Biomedicine laboratory, Biobank, CHRIS Center
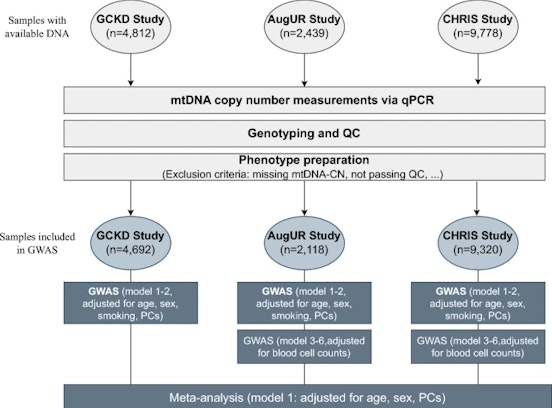
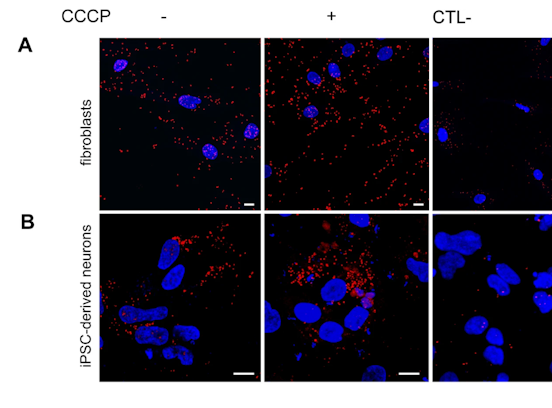
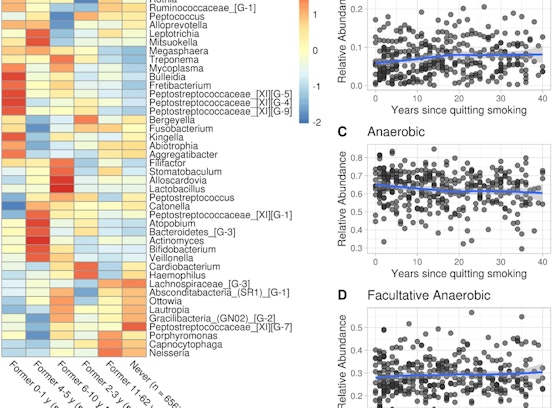
Data refer to the last issue of the Activity Report. See all Facts & Figures.
Research Field
1 - 3
News & Events
1 - 10